Pólipos e Polipose Intestinal: Diagnóstico e Tratamento
Cristiane Boé e Ulysses Fagundes Neto
Introdução
Por definição Pólipos são massas tumorais que se projetam em direção à luz intestinal. Presume-se que comecem como lesões pequenas e sésseis e, em muitos casos, devido a uma tração exercida sobre a superfície da massa pode criar uma haste, constituindo assim um pólipo pediculado.
Os pólipos são classificados segundo o quadro abaixo:
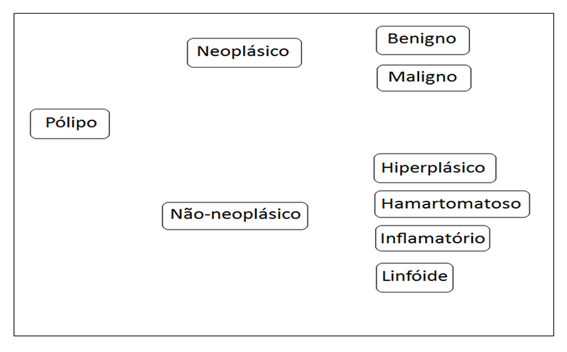
Os Pólipos epiteliais que surgem como resultado de proliferação e displasia são denominados Pólipos adenomatosos. São lesões verdadeiramente neoplásicas e precursoras de carcinomas. Por outro lado, Pólipos hamartomatosos são malformações das glândulas e do estroma; a lâmina própria constitui sua maior parte e, em geral não têm potencial de malignidade.
Pólipos Isolados
Os pólipos se tornam clinicamente relevantes quando causam sangramento retal, obstrução intestinal ou devido ao seu potencial de desenvolver malignidade.
Pólipos juvenis esporádicos são os mais comuns em crianças, são hamartomatosos e limitados ao cólon. Também chamados Pólipos de retenção ou inflamatórios, correspondem a aproximadamente 90% dos Pólipos colônicos em crianças. Sua apresentação mais comum é com sangramento intestinal baixo e dor abdominal. O sangramento causado geralmente é vermelho, indicando sua localização distal. É autolimitado e, frequentemente, há história de passagem de sangue após a eliminação das fezes.
Melena só é vista em casos raros de Pólipos gástricos ou duodenais, em especial, naqueles pacientes com síndromes de polipose intestinal.
No caso do Pólipo se projetar para a luz intestinal, ele pode, pela peristalse e tração, desenvolver intussucepção. Pode também se apresentar como um prolapso de massa retal, ou fezes mucopurulentas.
Pólipos juvenis são diagnosticados nos primeiros 10 anos de vida, com pico entre 2 e 5 anos, 50% das crianças com Pólipos juvenis têm mais de 1 pólipo. Os Pólipos costumam medir de 1 a 3 cm, e 90% deles são pediculados.
É importante ressaltar que Pólipos juvenis solitários não apresentam risco de se tornarem neoplasias malignas, porém, quando há mais que 5 Pólipos, existe o risco de desenvolver carcinoma colo-retal (Síndrome da Polipose Juvenil).
A colonoscopia com realização da polipectomia e sua respectiva revisão histológica são suficientes para o manejo dos Pólipos juvenis isolados. Não é necessário seguimento clínico ou endoscópico após a realização da polipectomia.
Histologicamente, o Pólipo juvenil típico tem arquitetura cística, glândulas mucoides, lâmina própria proeminente e denso infiltrado de células inflamatórias.
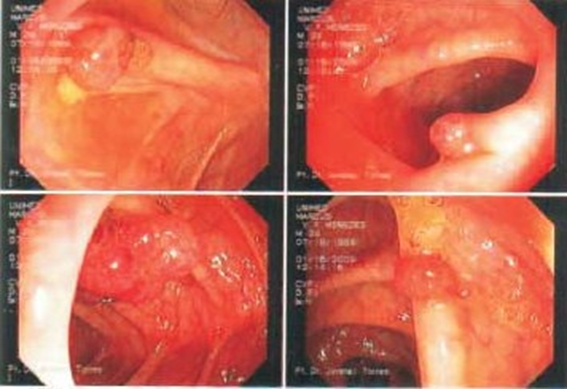
Colonoscopia – Pólipos sésseis e pediculados no ceco
Poliposes Intestinais
As Síndromes de Polipose Intestinal são relativamente raras na prática da gastroenterologia pediátrica, mesmo em serviços acadêmicos de referência. Entretanto, é importante que os especialistas estejam cientes dos riscos acarretados para as crianças e suas famílias afetados por estas patologias.
 As Síndromes das Poliposes hereditárias estão associadas a um risco maior de desenvolver câncer, por isso, uma classificação precisa delas é essencial. Certamente as lesões extraintestinais podem alertar os patologistas para a possibilidade de uma Síndrome de Polipose Intestinal.
As Síndromes das Poliposes hereditárias estão associadas a um risco maior de desenvolver câncer, por isso, uma classificação precisa delas é essencial. Certamente as lesões extraintestinais podem alertar os patologistas para a possibilidade de uma Síndrome de Polipose Intestinal.
Síndrome de Peutz-Jeghers
Trata-se de uma Síndrome de herança autossômica dominante e causada por uma mutação no gene STK11 (LKB1), caracterizada por múltiplos pólipos no trato gastrointestinal, associados a pigmentação mucocutânea, especialmente na borda dos lábios. Sua incidência é estimada entre 1:50.000 a 1:200.000.
Características clínicas




Os Pólipos podem ser encontrados por todo o trato gastrointestinal, mas a maioria está localizada no intestino delgado (60% a 94%) e no cólon (50% a 64%). Os Pólipos podem causar sangramento gastrointestinal, anemia e dor abdominal secundária à intussuscepção, obstrução ou infarto.
Os sintomas relacionados aos Pólipos são maiores na infância, 33% ocorrem até 10 anos de idade e 50% até 20 anos.
Um único indivíduo que apresente um dos seguintes achados:
1) dois ou mais Pólipos Juvenis confirmados histologicamente;
2) qualquer número de Pólipos Juvenis detectados em um indivíduo que tem história familiar de Síndrome de Peutz-Jeghers;
3) pigmentação mucocutânea em um indivíduo com história familiar de Síndrome de Peutz-Jeghers em parentes próximos;
4) qualquer número de Pólipos Juvenis em um indivíduo que também tenha pigmentação mucocutânea característica.
Não há correlação clínica clara demonstrada nos pacientes com mutação no gene STK1 e naqueles sem a mutação.
Durante o seguimento, existe a preocupação de detectar os Pólipos que possam causar invaginação intestinal/obstrução, sangramento/anemia.
Outra preocupação é a detecção de câncer nos estágios iniciais. O risco de câncer colorretal é 3%, 5%, 15% e 39% nas idades 40, 50, 60 e 70 anos, respectivamente, em pacientes com a Síndrome de Peutz-Jeghers.
A investigação do intestino delgado deve ser realizada para a prevenção de invaginação intestinal e necessidade de laparotomia de emergência. É realizada através da cápsula endoscópica. Outras alternativas são a ressonância magnética e a radiografia contrastada.
Tratamento
A polipectomia endoscópica reduz as complicações e o risco de uma futura polipectomia por laparotomia. Caso a laparotomia tenha que ser realizada (obstrução, invaginação ou retirada eletiva de grande quantidade de pólipos sintomáticos), a enteroscopia intra-operatória deve ser realizada com polipectomia, caso os Pólipos sejam detectados no intestino delgado (‘clean sweep’). Somente 40% das crianças deixam de necessitar a laparotomia na infância.
Não há tratamentos medicamentosos comprovados que possibilitem a redução da quantidade de Pólipos na Síndrome de Peutz-Jeghers.
Seguimento endoscópico
A colonoscopia é recomendada a cada 3 anos, a partir do início dos sintomas, ou na adolescência nos casos assintomáticos.
A endoscopia digestiva alta e o exame contrastado do trato gastrointestinal superior deve ser feita a cada 2 anos, a partir dos 10 anos de idade.
Os Pólipos, sempre que possível, devem ser removidos.
Polipose Juvenil
Esta síndrome caracteriza-se pelo desenvolvimento de múltiplos Pólipos juvenis no trato gastrointestinal.
Os sintomas costumam aparecer antes dos 20 anos de idade, e apresenta incidência aproximada de 1:100.000 indivíduos, diferentemente dos Pólipos juvenis isolados cuja incidência é de 2% em crianças.
A síndrome apresenta herança autossômica dominante, ocorre uma mutação e desregulação na inibição do fator de crescimento TGFβ levando aos múltiplos Pólipos gastrointestinais.
Muitos pacientes são diagnosticados tardiamente, pois não houve distinção entre Polipose Juvenil e Pólipos Juvenis esporádicos.
Características clínicas
Geralmente são encontrados múltiplos Pólipos no cólon, embora Pólipos gástricos e no intestino delgado também sejam observados.
Os pacientes podem apresentar prolapso retal e anemia, hipoproteinemia, malnutrição e distúrbios hidroeletrolíticos também podem ser encontrados.
A localização e o número de Pólipos variam muito, portanto, o tratamento endoscópico e cirúrgico deve ser individualizado.
Critérios diagnósticos
Devem ser observados os seguintes achados:
a- mais de 3 a 5 Pólipos juvenis colorretais;
b- Pólipos juvenis ao longo de todo o trato gastrointestinal;
c- qualquer número de Pólipos e história familiar de Polipose Juvenil.
Deve-se considerar que esta definição é problemática, visto que é relativamente comum encontrarmos crianças com múltiplos Pólipos juvenis (3 a 10 ou mais), sem história familiar de Polipose Juvenil.


Seguimento endoscópico
A colonoscopia e a endoscopia digestiva alta bienais ou trienais são recomendadas a partir dos 15 anos de idade ou antes, caso os Pólipos sejam clinicamente aparentes. A polipectomia deve ser realizada sempre que possível por via endoscópica, porém no caso de não ser possível a utilização desta técnica a cirurgia está indicada.
Considerando-se que a Polipose Juvenil é uma síndrome rara e a neoplasia colorretal na faixa etária pediátrica é extremamente incomum, as evidências para o seguimento endoscópico são limitadas.
Síndrome de Cowden
Trata-se de uma enfermidade rara, os pacientes manifestam hamartomas dos 3 tipos de tecidos: ectoderma, mesoderma e endoderma.
Do ponto de vista genético é de transmissão autossômica dominante, 85% dos pacientes apresentam mutação no gene supressor de tumor PTEN.
A enfermidade costuma se apresentar na adolescência e em adultos jovens, com prevalência estimada em 1: 200.000 a 250.000.
Manifestações clínicas
Papilomas hiperqueratóticos nos lábios, língua e narinas são os achados extraintestinais mais comuns. Os marcadores mais importantes da síndrome são os achados mucocutâneos. Os achados patognomônicos são: trichilemmomas (pápulas verrucóides assintomáticas), papilomas nas cavidades mucosas, queratose acral nas mãos e pés, doença de Lhermitte-Duclos (gangliocitoma displásico do cerebelo), os quais são menos frequentes na infância que em adultos.




Achados benígnos: lipomas, hemangiomas e neuromas, macrocefalia, adenomas e lesões fibrocísticas das mamas, adenomas de tireóide, leiomiomas uterinos (são critérios menores).
Pólipos hamartomatosos ocorrem por todo trato gastrointestinal, geralmente assintomáticos. Pólipos juvenis são os mais comuns, porém também aparecem outros tipos como lipoma, pólipos inflamatórios, ganglioneuromas, hiperplasia linfóide, e adenomas.
Síndrome Ruvalcaba-Myhre-Smith
Trata-se de uma síndrome de herança autossômica dominante, também associada com mutação no gene PTEN (inibidor de crescimento celular). Admite-se que esta enfermidade pode ser uma variação da Síndrome de Cowden.
Até o presente momento não há um consenso sobre os critérios diagnósticos desta síndrome, porém, é sabido que indivíduos apresentando várias combinações de macrocefalia, lipomatose, hemangioma, pólipos gastrointestinais e máculas pigmentadas na glande do pênis, são considerados clinicamente afetados. Atraso mental e outras anomalias congênitas também são associados com a síndrome. Acredita-se que o câncer de mama, tireoide e endométrio sejam componentes da síndrome.
Em contraste com a Síndrome de Cowden, os Pólipos são frequentemente sintomáticos, juvenis, encontrados no íleo e cólon.
Seguimento endoscópico
Não existem recomendações baseadas em evidências para a vigilância do trato gastrointestinal em crianças ou adultos que possuem diagnóstico de síndromes hamartomatosas com mutação PTEN.
Polipose Adenomatosa Familiar (PAF)
Trata-se da polipose mais comum, sua prevalência é estimada em 1:5000 a 1:17000. Esta lesão é causada por mutação no gene supressor de tumor APC. Vale ressaltar que a maioria dos pacientes pediátricos com PAF são assintomáticos, e a evolução é baseada na história familiar.
Pólipos colônicos benignos costumam aparecer em torno dos 16 anos, sua principal característica é o desenvolvimento de grande número de Pólipos colorretais, os quais são adenomatosos e inevitavelmente evoluem para câncer colorretal.
Em associação com os Pólipos gastrointestinais, há numerosas manifestações extraintestinais da PAF aumentando o risco de câncer cerebral, tireoide, hepático e pancreático. Em particular, o risco de hepatoblastoma em pacientes com PAF é 850 vezes maior que na população em geral.
Diagnóstico
Baseia-se no achado de mais de 100 pólipos adenomatosos no cólon, sendo que a maioria dos pacientes têm entre 100 e 1000 Pólipos distribuídos por todo o cólon.
A existência de poucos adenomas sugere polipose atenuada (AFAP) ou defeito no gene MYH.
Manifestações clínicas
Alkhouri et al. estudaram 12 pacientes com PAF de 1990 a 2005. As idades variaram de 7 a 18 anos, e a queixa principal, sangramento retal como manifestação inicial, estava presente em 86% dos casos. Outros achados foram dor abdominal e anemia comprovada por exame laboratorial.
Deste grupo, 1 paciente de 8 anos apresentava 3 pólipos na colonoscopia inicial e após 1 ano mais de 200; 1 paciente foi diagnosticado com câncer colorretal in situ aos 18 anos;outros pacientes apresentavam no início do acompanhamento entre 8 e 200 pólipos.
O risco de desenvolvimento de câncer antes de 21 anos varia de 0,21% a 7%.
Em virtude do risco de adenocarcinoma de duodeno e considerando a ocorrência de uma grande proporção de pacientes com lesões duodenais, a realização da endoscopia digestiva alta é recomendada no momento da colonoscopia inicial. Sabe-se que 45% das crianças apresentam adenomas gástricos e duodenais.
Para a investigação do intestino delgado, pode ser realizado RX contrastado ou, mais recentemente, utilizar-se da cápsula endoscópica. Uma outra opção factível de investigação é a realização da Ressonância Magnética.
Uma vez diagnosticada a PAF, a indicação cirúrgica é o único tratamento reconhecido para redução do risco de câncer colorretal. O momento de ser realizada a colectomia é decidido de paciente para paciente. O mais recomendado é que se aguarde a adolescência para que o próprio possa participar mais ativamente desta decisão.
Entretanto, vale enfatizar que o risco de desenvolver câncer de intestino delgado permanece o mesmo após a colectomia.
Sulindac é um antiinflamatório não-esteroidal que têm se mostrado efetivo na redução do número de adenomas colorretais já estabelecidos em aproximadamente 50%, entretanto, não previne o desenvolvimento de adenomas primários.
Celecoxib, inibidor seletivo da Cox-2, têm se mostrado promissor na redução do número de adenomas colorretais e duodenais. Estudo randomizado, duplo-cego em fase III está sendo conduzido para testar quando este medicamento pode ser usado para prevenir a formação de Pólipos colorretais em crianças com FAP.
Seguimento
Teste genético é recomendado para pacientes com risco de desenvolver FAP, entre 10 e 12 anos de idade, quando também é sugerida a primeira endoscopia.
Colonoscopia bienal deve ser realizada em crianças de risco aos 10 e 12 anos, a menos que exista doença agressiva em familiares.
Uma vez que os adenomas sejam identificados, é geralmente recomendada colectomia com anastomose íleo-anal.



Síndrome de Gardner


Tem sido descrita hipertrofia congênita do epitélio pigmentar da retina e as neoplasias associadas são carcinomas da ampola de Vater, adrenal e tireoide.
Os sintomas costumam aparecer entre os 2 meses e os 20 anos de idade. Geralmente as manifestações extracolônicas, como os tumores de pele e osteomas surgem antes dos Pólipos adenomatosos. As lesões de pele são cistos sebáceos, lipomas, fibromas e lesões pigmentadas.
Os riscos de desenvolvimento de câncer colorretal são os mesmos da PAF.
Síndrome de Turcot
Foi descrita por Jacques Turcot originalmente caracterizada por Pólipos hamartomatosos e tumores de sistema nervoso central. No entanto, o autor também descreveu câncer colorretal hereditário não-polipóide, secundário a uma mutação num gene de reparo do DNA, associado a tumor primário do cérebro (glioblastoma cerebelar).
Quando os Pólipos colorretais são secundários à mutação no gene APC, o tumor primário do cérebro geralmente é um meduloblastoma cerebelar. Estima-se que 2/3 dos casos com mutação no gene APC e múltiplos Pólipos, apresentarão meduloblastoma.
Síndrome de Cronkhite-Canada
Esta é uma síndrome não-hereditária de Pólipos juvenis em associação com anormalidades ectodérmicas, tais como, alopécia, distrofia ungueal, hiperpigmentação da face e pálpebras, diarreia e perda de peso.
A patogênese desta síndrome rara (400 casos relatados até hoje, 75% no Japão) é desconhecida e o manejo é apenas suporte clínico.
Em uma revisão da literatura, 34 de 280 casos tiveram associação com câncer colorretal.
Hiperplasia Nodular Linfoide (HNL)
A HNL caracteriza-se pela existência de hiperplasias dos folículos linfoides do cólon, os quais costumam ser pouco comuns e são de natureza benigna.
O tratamento para este tipo de Pólipo é sua retirada e análise histopatológica, para diferenciação com linfoma de cólon.
Polipose Inflamatória
São vistos em associação com diversas condições inflamatórias, incluindo a Doença Inflamatória Intestinal. Podem ser referidos como pseudopólipos.
Estes Pólipos se desenvolvem como resultante de ciclos repetidos de inflamação e cicatrização em pacientes com doença inflamatória crônica. Estão geralmente confinados ao cólon.
Ocorrem em aproximadamente 1/3 dos pacientes com Doença de Crohn, porém outras condições associadas são colite isquêmica, infecciosa e Doença de Behçet.
Conclusão
O capítulo dos Pólipos intestinais reveste-se de extrema importância clínica, pois além de ser vasto, envolve uma série de preocupações diagnósticas diferenciais quanto a sua natureza, que pode ser benigna ou maligna, o que acarreta a necessidade de uma investigação profunda e criteriosa para que se possa oferecer a melhor conduta terapêutica para nossos pacientes.