Insuficiência Pancreática: Etiologia e Tratamento
Insuficiência Pancreática: Etiologia e Tratamento
Cristiane Boé e Ulysses Fagundes Neto
Introdução
A função pancreática normal garante efetiva digestão e absorção dos nutrientes. Vale ressaltar que o pâncreas adquire plena maturidade funcional na produção enzimática a partir dos 2 anos de idade (Figuras 1-2-3).
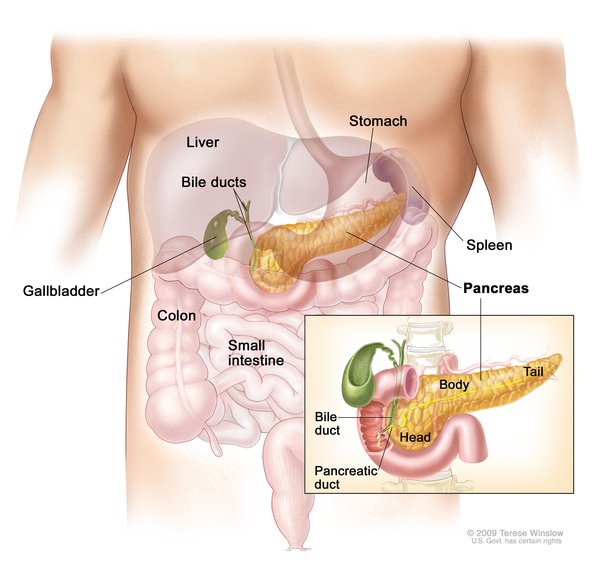
Figura 1- Localização anatômica do pâncreas no interior da cavidade abdominal.
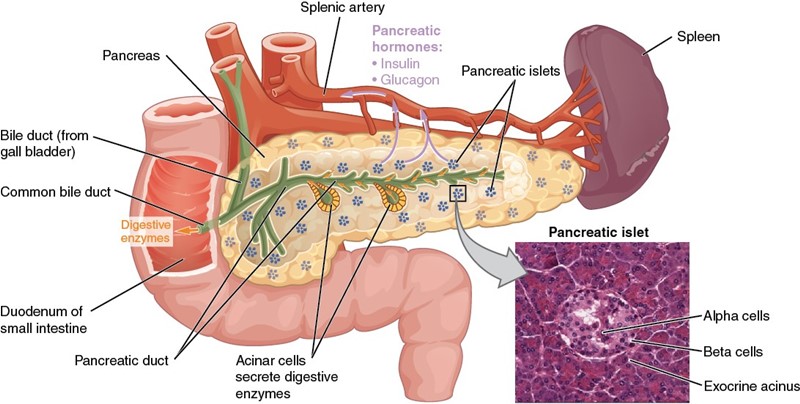
Figura 2- Anatomia e histologia do pâncreas e sua localização na segunda porção do duodeno.
Por outro lado, as manifestações clínicas da insuficiência pancreática (IP) ocorrem quando as secreções exócrinas do pâncreas não mantêm uma função digestiva normal, afetando a qualidade de vida e, como consequência, pode resultar em desnutrição.
A principal causa de IP é a pancreatite crônica, que apresenta a estimativa de afetar 0,4% a 5% da população mundial. Entretanto, em crianças, a causa mais comum é a fibrose cística (FC). A prevalência de IP exócrina nos casos de pancreatite crônica é de 30 a 40%, enquanto que na FC é de 80 a 90% (Figura C).
Desde 1938, após a aprovação pelo Food and Drug Administration (FDA), a reposição de enzimas pancreáticos tem sido a principal ação terapêutica para aqueles pacientes que sofrem de má absorção dos nutrientes secundária à insuficiência pancreática. O nível das enzimas pancreáticas secretadas no intestino delgado varia segundo a quantidade e o tipo de lipídios ingeridos durante uma refeição, além de também apresentar uma oscilação individual. Em virtude da existência destas variáveis, determinar o nível exato de enzimas secretadas por um indivíduo normal é muito difícil.
Etiologias
As causas de IP em crianças podem ser divididas em 2 categorias:
– hereditárias
– adquiridas
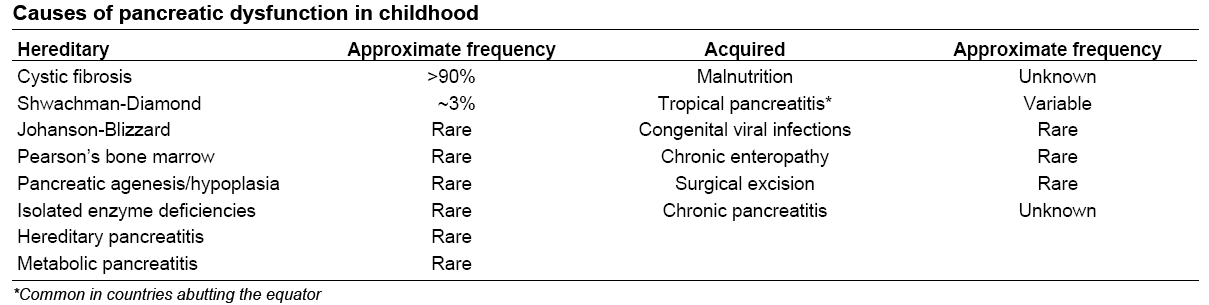
Quadro 1- Causas de IP na infância.
A maioria das causas hereditárias faz parte de doenças sistêmicas, como explicitado no quadro 1, abaixo.
Apresentação Clínica da Insuficiência Panceática
O diagnóstico é em grande essência clínico. No caso de um determinado paciente ser portador de alguma causa conhecida de IP que se apresenta com perda de peso e esteatorreia, está indicado o início do tratamento sem que seja necessária a realização de extensa investigação laboratorial. A IP se instala quando restar menos de 10% da função pancreática exócrina.
Esteatorreia (fezes espumantes, malcheirosas que boiam no vaso sanitário), perda de peso, desconforto e distensão abdominal são sintomas comuns relacionados com a digestão inadequada dos lipídios.
Inicialmente, a digestão de proteínas e amido se mantém adequada. Com a progressão da IP a má absorção de lipídios leva ao quadro de desnutrição, má absorção de vitaminas lipossolúveis (A, D, E, K), depleção de micronutrientes e diminuição de lipoproteínas circulantes.
A IP exócrina pode por si só, de forma isolada, causar ou exacerbar transtornos da motilidade do trato digestivo. Nestas circunstâncias, há alteração na regulação neuro-hormonal, especificamente na produção de colecisticinina e polipeptídeos pancreáticos (afetados pelo alimento não digerido no intestino), levando ao esvaziamento rápido do estômago e alterando a motilidade antroduodenal e da vesícula biliar.
Nos pacientes, nos quais o tratamento ainda não se iniciou, é notório o fato de que se alimentam em curtos espaços e que apresentam tempo de trânsito no intestino delgado mais rápido, cujos sintomas podem ser revertidos com o emprego de enzimas pancreáticos exógenos.
Diagnóstico
A caracterização diagnóstica pode ser estabelecida por métodos diretos e indiretos, a saber:
strong>Indiretos: são menos invasivos, mas que apresentam alta sensibilidade e especificidade, tais como:
– determinação quantitativa da eliminação de gordura nas fezes, as quais devem ser coletadas por um período de 72 horas, pelo método de Van de Kamer;
– elastase fecal, dosada pela técnica ELISA, e que avalia exclusivamente a produção enzimática endógena;
– quimiotripsina fecal, determinada por método colorimétrico, avalia a produção de ambas enzimas, endógenas e exógenas, portanto, pode apresentar resultados duvidosos em pacientes em vigência de terapia de reposição enzimática.
Diretos (quantitativos)
– teste da secretina-ceruleina;
– teste da secretina-pancreozimina (padrão-ouro, porém invasivo, sem valores de referência bem definidos em crianças).
- Teste da secretina-pancreozimina
Deve-se utilizar sonda gastroduodenal de dupla via posicionada sob controle fluoroscópico; uma das vias drena o conteúdo gástrico e a outra o duodenal. Secreções duodenais são coletadas em intervalos de 10 minutos, em três períodos após injeção intravenosa de secretina e em três períodos após infusão intravenosa de pancreozimina. Os aspirados são avaliados quanto ao volume, medida do pH, concentração de bicarbonato, amilase, tripsina, lipase, bilirrubinas, fosfatase alcalina, cálcio e magnésio. Amostras são encaminhadas para microscopia e citologia. A ocorrência de produção enzimática reduzida após injeção de pancreozimina corresponde ao índice mais sensível para avaliar a diminuição da função pancreática.
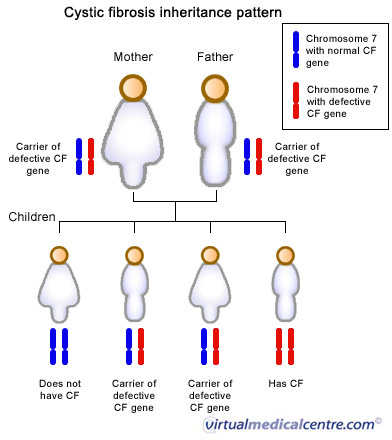
Figura 3- Tipo de herança da FC.
Principais Enfermidades Causadoras de Insuficiência Pancreática
Fibrose Cística
Trata-se de doença de herança autossômica recessiva que afeta múltiplos tecidos secretores epiteliais cuja incidência é de 1:3000 nascidos vivos em indivíduos caucasianos (Figura 3).
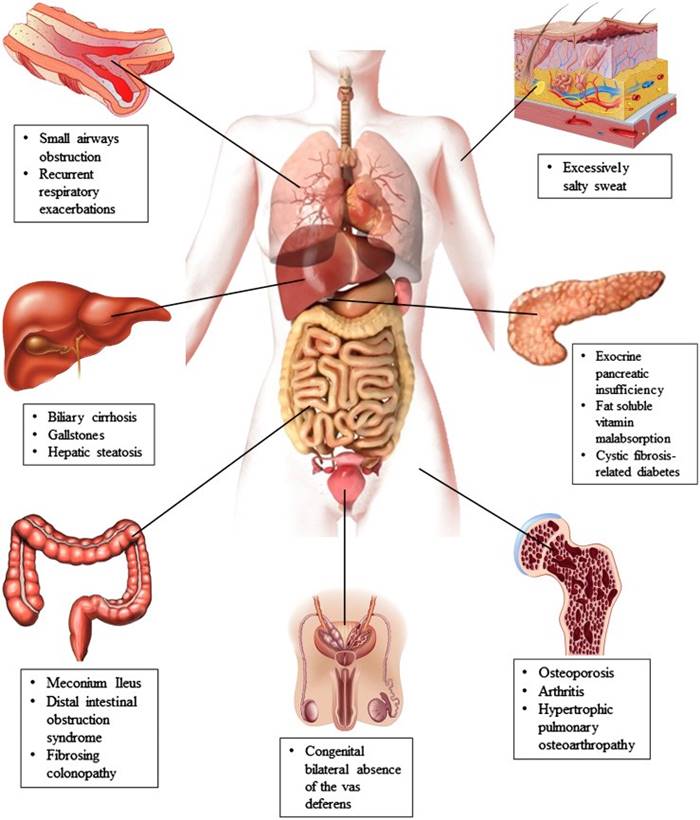
Figura 4- Principais manifestações clínicas e órgãos afetados pela FC.
As manifestações clínicas da FC são reflexo de mutações no gene CFTR (cystic fibrosis transmembrane condutance regulator). Geralmente ocorrem 2 mutações (forma clássica) ou 1 mutação (forma não-clássica), sendo a mais frequente aquela que afeta o gene ΔF508. Estas mutações no gene CFTR resultam em transporte anormal de sódio e cloro. Em condições normais, o cloro intraluminal é trocado por bicarbonato, o que torna o ambiente alcalino, permitindo, desta forma, que altas concentrações de proteínas fiquem em seu estado solúvel. Em virtude da mutação no gene, o resultado é a presença de uma secreção viscosa e ácida, levando à obstrução ductal. Esta obstrução resulta em destruição tecidual por retenção de enzimas proteolíticas, fibrose, substituição gordurosa, formação de cistos e insuficiência pancreática exócrina.
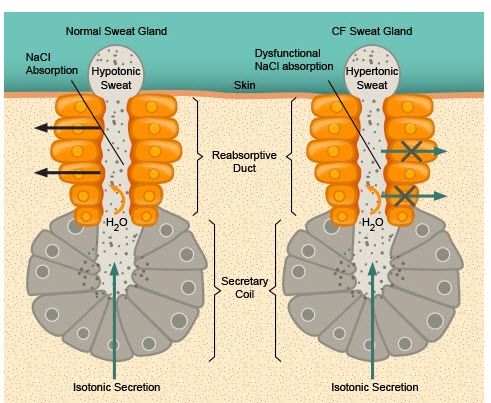
Figura 5- Esquema representativo comparando a glândula sudorípara de um indivíduo normal com a do portador de FC.
O mesmo ocorre com as secreções pulmonares (doença pulmonar crônica com infecções bacterianas recorrentes) e infertilidade no sexo masculino devido a azoospermia obstrutiva.
Nos recém-nascidos deve-se suspeitar do diagnóstico de FC na presença de íleo meconial, prolapso retal, “suor salgado”, icterícia prolongada, anemia hemolítica sem causa aparente, hipoalbuminemia e edema.
Nas crianças maiores a existência de dedos em baquetas de tambor, pólipos nasais, assim como secreção do trato respiratório colonizada por S. aureus ou P. aeruginosa, são achados importantes.
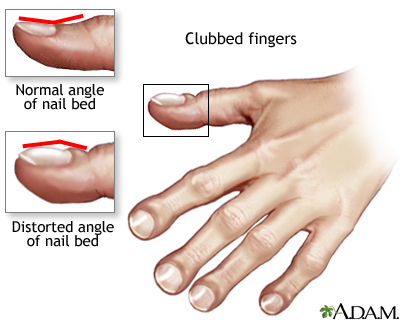
Figura 7- Dedos em baqueta de tambor causados pela FC de longa duração.
Triagem Neonatal
Desde 2001 o Ministério da Saúde criou o Programa Nacional de Triagem Neonatal, incluindo o diagnóstico precoce para FC, anemia falciforme e outras hemoglobinopatias.
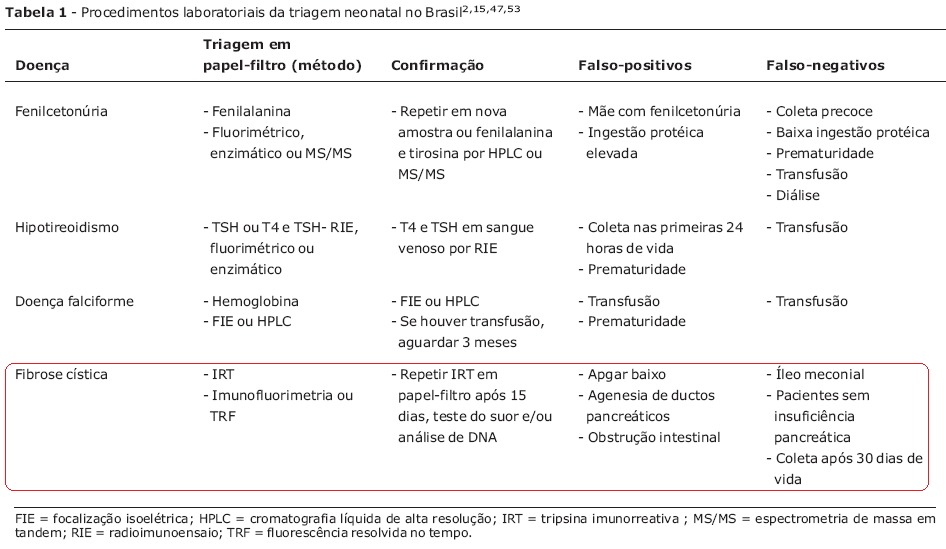
Realiza-se rotineiramente a dosagem sérica do tripsinogênio imunorreativo (IRT), indicador indireto da doença pois avalia a integridade da função pancreática (Tabela 1).
Caso a dosagem revele-se positiva (>70ng/ml), deve-se realizar nova dosagem após 15 dias, e se o teste permanecer positivo, deve-se realizar teste genético ou teste do suor.
O Íleo meconial pode estar relacionado a testes falso‐negativos, pois com a desobstrução intestinal ocorre rápida queda dos níveis de IRT no sangue. Mesmo diante de um IRT normal, o que não descarta completamente o diagnóstico de FC, se uma criança apresentar sintomas sugestivos da doença, deve ser submetida ao teste do suor.
Figura 8- Representação esquemática das provas laboratoriais para a confirmação diagnóstica de FC desde o nascimento.
Diagnóstico
O diagnóstico de FC deve ser suspeitado na presença de uma ou mais manifestações fenotípicas (clínicas) características:
- Doença sinusal ou pulmonar crônica e/ou;
- Insuficiência exócrina pancreática crônica e/ou;
- História familiar de FC confirmada e/ou;
- Teste duplamente positivo de triagem neonatal.
Associada(s) à evidência de elevação anormal da concentração de Cloro no suor, em duas ocasiões diferentes ou, em casos especiais, deve-se solicitar a identificação genética de duas mutações da FC.

Figura 9- Esquema do procedimento do teste do suor.
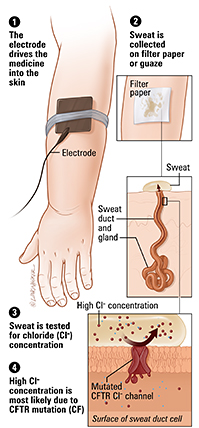
Figura 10- Representação esquemática dos princípios que se baseia o teste do suor.
O teste do suor realizado pelo teste da iontoforese de pilocarpina é o método mais sensível para o diagnóstico da FC. As seguintes definições são recomendadas para a interpretação do teste do suor:
– concentração de Cloro >60 mmol/l dá suporte para o diagnóstico de FC;
– concentração intermediária de Cloro entre 40‐60 mmol/l é sugestivo, porém não diagnóstico;
– concentração de Cloro <40 mmol/l é normal;
– a concentração de Sódio não deve ser interpretada na ausência da respectiva dosagem do Cloro;
– condutividade <60mmol/l é improvável estar associada a FC e valores >90mmol/l são sugestivos de FC.
Tratamento da insuficiência pancreática exócrina
Recomenda-se a terapia de substituição enzimática, de acordo com cada caso (pancreolipase). A dose de enzimas baseia-se no peso do paciente e deve começar com 1000 unidades de lipase/kg/refeição para crianças com menos de 4 anos de idade, e com 500 unidades de lipase/kg/refeição para crianças com mais de 4 anos; a dose deve ser ajustada de acordo com a gravidade da doença, o controle da esteatorréia e a manutenção de um bom estado nutricional; de um modo geral, os pacientes não devem exceder a dose de 10.000 unidades de lipase/kg de peso por dia.
As enzimas pancreáticas exógenas devem ser ingeridas antes de cada refeição. A adequação da dose das enzimas ingeridas deve ser realizada através da dosagem da gordura fecal coletadas durante 72h (Van de Kamer), além da avaliação clínica levando-se em consideração a curva ponderal e a ocorrência ou não de diarreia.
A elastase fecal e a quimiotripsina fecal servem para avaliar a função pancreática, mas não para ajustar a dose de enzimas.
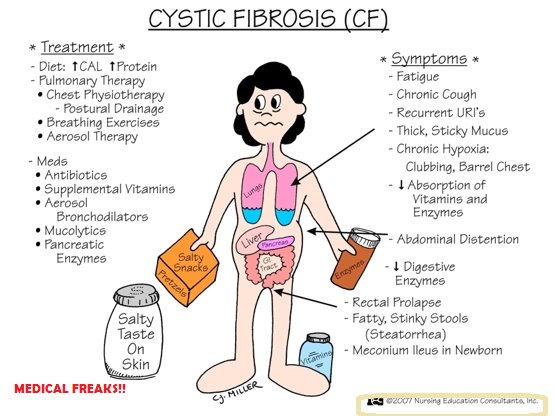
Figura 11- Princípios básicos do tratamento da FC.
Colonopatia fibrosante:
Trata-se da complicação causada pelo uso crônico de altas doses de enzimas pancreáticas para o controle da má absorção. A dose de enzima associada a esta complicação varia de 24.000 UI a 50.000 UI de lipase/kg/dia. Como resultado, há deposição de colágeno na submucosa do intestino grosso, com consequente fibrose.
Fracasso no tratamento:
No caso de haver um fracasso terapêutico, como primeiro passo deve-se avaliar a aderência ao tratamento, à dieta e à hora de administração da enzima. A primeira opção é aumentar a dose de lipase oferecida e observar a resposta. Caso não houver sucesso, recomenda-se modificar a dieta, em particular reduzindo o consumo de gordura para 50 a 75 g/dia. Deve-se também reduzir o consumo de fibras, álcool, cálcio e magnésio, pois estas substâncias podem contribuir para aumentar a esteatorreia. Quando necessário o próximo passo é administrar medicamentos anti-ácidos, como inibidores do H2 ou inibidores da bomba de prótons. Caso não ocorra diminuição da esteatorreia, é recomendada a investigação de possível infestação por Giardia e, também, a existência de sobrecrescimento bacteriano no intestino delgado.
Síndrome Shwachman-Diamond (SSD)
Esta enfermidade foi descrita primeiramente em 1964 e, seguindo a FC, é a causa mais comum de insuficiência pancreática exócrina em crianças. Sua etiologia é desconhecida, porém, tudo indica ser de herança autossômica recessiva. A insuficiência pancreática apresenta gravidade variável e é um achado consistente da síndrome. A lesão pancreática é devida a uma falha do desenvolvimento acinar do pâncreas. Nesta síndrome ocorre um extenso remodelamento gorduroso do tecido do pâncreas com arquitetura ductular normal.
Mais de 98% da capacidade acinar exócrina do pâncreas deve estar comprometida para que os sintomas da insuficiência pancreática apareçam. Usualmente a esteatorréia surge nos 6 primeiros meses de vida em 50% dos casos, e em 90% antes de 1 ano de vida. Níveis baixos de tripsinogênio pancreático e isoamilase são marcadores úteis de insuficiência pancreática em pacientes com SSD. Em contraste com a FC, a função pancreática frequentemente melhora com a idade.
Excluir o diagnóstico de FC pelo teste do suor é essencial. O IRT na SSD deve ser normal ou baixo, pois também em contraste com a FC, a insuficiência pancreática é secundária à hipoplasia, não à obstrução/destruição do pâncreas.
A medida da elastase fecal é um bom método para triagem. A Tomografia Computadorizada, a Ressonância Magnética e a Ultrassonografia frequentemente mostram um pâncreas pequeno, com estrutura anormal e composto principalmente por gordura.
Hepatomegalia é encontrada em 15% dos casos e entre 50% e 75% deles apresentam enzimas de avaliação da função hepática em níveis de 2 a 3 vezes acima do valor da normalidade, isto se deve à esteatose hepática.
Além das manifestações de insuficiência pancreática também podem ocorrer inúmeras outras anormalidades extra-pancreáticas conforme as que estão descritas na Figura 12 e no Quadro 2, abaixo.

Quadro 3- Critérios diagnósticos da SSD.
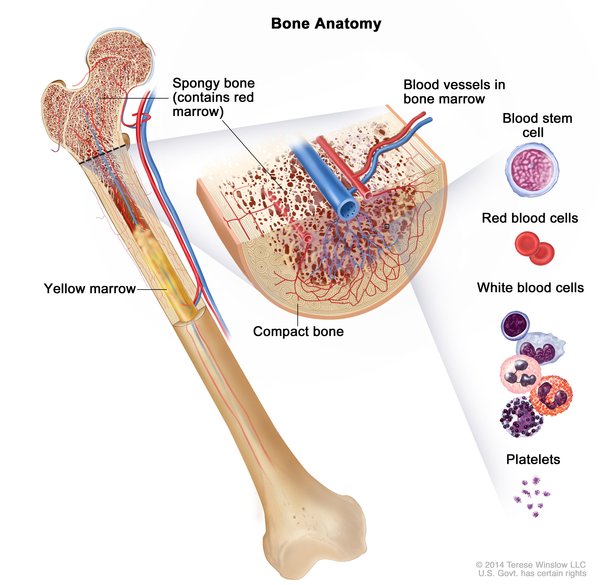
Figura 12- Principais repercussões clínicas da SSD.
O Quadro 3 apresenta um esquema dos critérios diagnósticos da SSD.
A maioria dos pacientes requer suplementação de enzimas pancreáticas, entretanto, a esteatorreia desaparece espontaneamente em aproximadamente 50% dos casos.
A reposição de vitaminas lipossolúveis está indicada (A, D, E e K). O tempo de protrombina quando está anormal pode ser útil como marcador para deficiência de vitamina K.
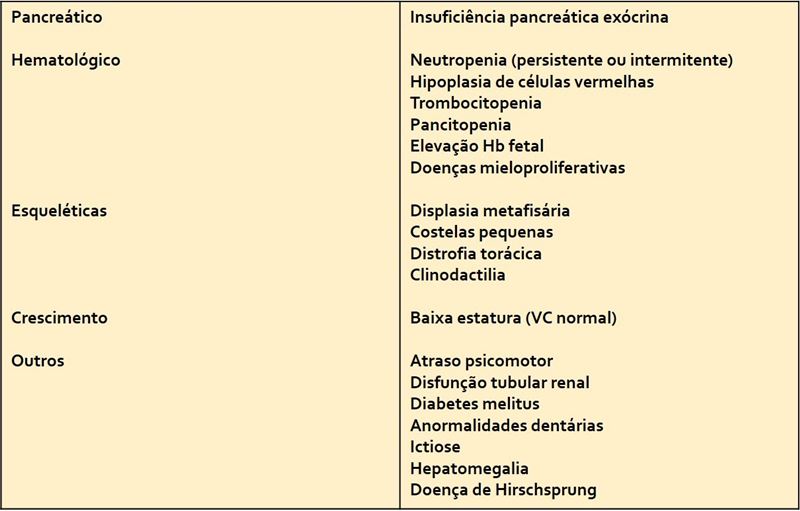
Quadro 2- Relação das diversas possíveis manifestações extra-pancreáticas na SSD.
Síndrome Johanson-Blizzard
Trata-se de uma síndrome rara, multissistêmica de herança autossômica recessiva, decorrente de mutação no gene UBR1 no cromossomo 15q15-21, descrita pela primeira vez em 1971. Até 2002, tinham sido relatados 38 pacientes com esta síndrome, sendo a insuficiência pancreática um achado constante em 37 deles. Outros achados frequentes são: aplasia da aleta nasal, surdez, hipotireoidismo, ausência de dentes permanentes, aplasia do couro cabeludo e insuficiência pancreática exócrina (Tabela 1).
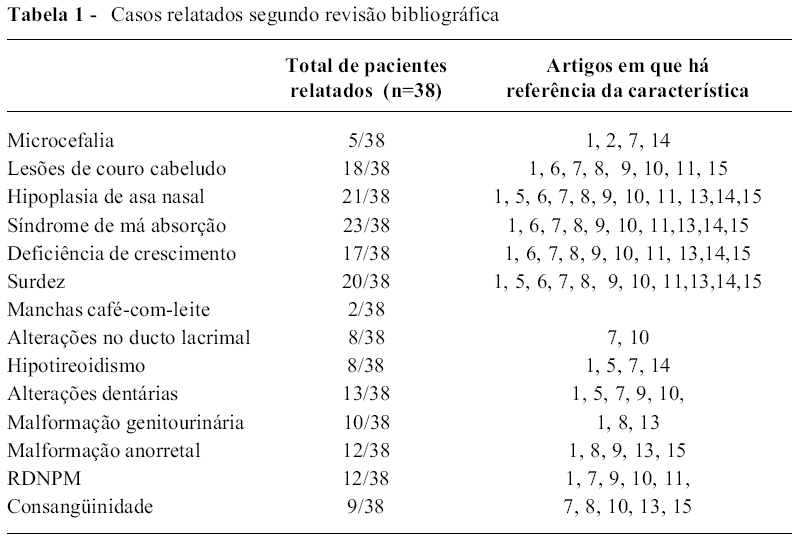
A agenesia da cartilagem nasal torna o diagnóstico da síndrome mais fácil, bem como a ausência de anomalias na medula óssea e esqueléticas a diferencia da SSD. A insuficiência pancreática é devida a defeitos primários das células acinares, similar ao que ocorre na SSD. A Tomografia Computadorizada do abdome mostra remodelamento gorduroso do pâncreas. Vale ressaltar que há descrição de evolução para insuficiência pancreática endócrina (Diabetes mellitus).
É importante enfatizar que nos lactentes a morte pode ocorrer em virtude de má absorção grave secundária ao quadro de insuficiência pancreática.
O tratamento é feito com reposição de enzimas pancreáticas.
Síndrome de Pearson
Trata-se de síndrome rara, caracterizada por insuficiência pancreática exócrina associada a anemia sideroblástica refratária ao tratamento. Logo após o nascimento, há anemia macrocítica com graus variáveis de neutropenia e trombocitopenia. O aspirado da medula óssea mostra vacuolização marcante dos precursores eritróides e mieloides, juntamente com grau intenso de sideroblastos em anel e hemossiderose, diferente dos achados descritos na SSD.
Os pacientes evoluem com déficit de crescimento e requerem transfusões sanguíneas frequentes. Os testes da função qualitativa pancreática revelam depressão da função acinar (análises de autópsias mostram atrofia das células acinares e fibrose).
O tratamento da disfunção pancreática também é realizado com reposição enzimática.
Conclusão
As causas de insuficiência pancreática podem ser de natureza variável e muito embora os casos de pancreatite aguda e/ou crônica sejam prevalentes na vida adulta, por outro lado, na infância as principais causas são de origem hereditária, e, portanto, devem chamar a atenção do clínico desde o período neonatal e ao longo do primeiro ano de vida. Deficiência do ganho pondero-estatural associada a diarreia com esteatorreia são as manifestações clínicas mais importantes e que devem levar a suspeição de insuficiência pancreática. Nestas circunstâncias impõem-se que a investigação diagnóstica seja realizada o mais rapidamente possível, visando proporcionar que o tratamento de reposição enzimática seja instituído de forma eficaz, para evitar a evolução para desnutrição e risco de vida.